
大家都知道美國FDA醫療器械分為3類,那它是怎么區分類別的呢?
美國FDA醫療器械分類方法,是所有出口美國企業必須面臨的問題。
美國FDA醫療器械產品目錄中共有1700多種。根據風險等級的不同,FDA將醫療器械分為三類(Ⅰ,Ⅱ,Ⅲ)。風險等級逐級升高,Ⅲ類風險等級高。
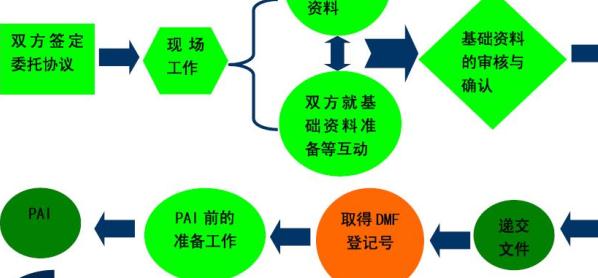
FDA對每一種醫療器械都明確規定了其產品分類和管理要求。首先,對于任何產品,企業都需進行企業注冊與產品列名。
I 類器械一般控制(General Control) 絕大部分產品只需進行注冊、列名和實施GMP規范。

I 類器械一般控制(General Control)絕大部分產品只需進行注冊、列名和實施GMP規范,產品即可進入美國市場(其中極少數產品連GMP也豁免,極少數保留產品則需向FDA遞交510(K)申請
這些器材只要經過一般控制就可以確保其功效與安全性,如拐杖、眼鏡片、膠布等,約占全部醫療器材的27%。
這些控制包括:禁止粗制濫造及不當標示的產品銷售;FDA得禁止不合格產品銷售必須報告FDA有關危害性、修理、置換等事項;限制某些器材的販賣、銷售、及使用;實施GMP
Ⅱ類產品(占46%左右)一般控制 + 特殊控制企業在進行注冊和列名后,這些產品除了上述一般控制之外,其余大多數產品均要求進行上市前通告。
少量的II類產品可以豁免上市前通告程序。生產企業須在產品上市前90天向FDA提出申請,通過510K審查后,產品才能夠上市銷售。
Ⅲ類產品(占7%左右)
一般控制 + 上市前許可企業在進行注冊和列名后,須實施GMP并向FDA認證遞交PMA申請(部分Ⅲ類產品還是PMN)。
一般來說, III類產品多為維持、支持生命或植入體內的器材,對病患具有潛在危險,可能引起傷害或疾病者,如心律調節器、子宮內器材及嬰兒保溫箱等,約占所有器材的8%。這些器材必須取得FDA的PMA之后方能銷售。
對Ⅰ類產品,企業向FDA遞交相關資料后,FDA只進行公告,并無相關證件發給企業;
對Ⅱ、Ⅲ類器械,企業須遞交PMN或PMA,FDA在公告的同時,會給企業以正式的市場準入批準函件(Clearance),即允許企業以自己的名義在美國醫療器械市場上直接銷售其產品。
至于申請過程中是否到企業進行現場GMP考核,則由FDA根據產品風險等級、管理要求和市場反饋等綜合因素決定。
綜合以上內容可知,絕大部分產品在進行企業注冊、產品列名和實施GMP,或再遞交510(K)申請后,即可獲得FDA批準上市。
本公司以責任為基礎,打造踏實肯干、拼搏不息的工作團隊,以誠信為根本,打造雙方互贏互利的經營模式,創新管理,完善技術、優化服務,打造良好的企業環境,創造“鋼鐵”一般的品質。

